Cleanroom Validation & Qualification: Essential for Contamination Control in 2025
Effective contamination control starts with robust cleanroom validation and qualification. With the full implementation of the EU GMP Annex 1 revision since August 2023, pharmaceutical manufacturers face expanded requirements for sterile drug production. The 2022 update, replacing the 2008 version, emphasizes clear guidance on premises, equipment, utilities, and personnel—helping you ensure regulatory compliance while maintaining sterility.
But why is this so critical for your contamination control strategy?
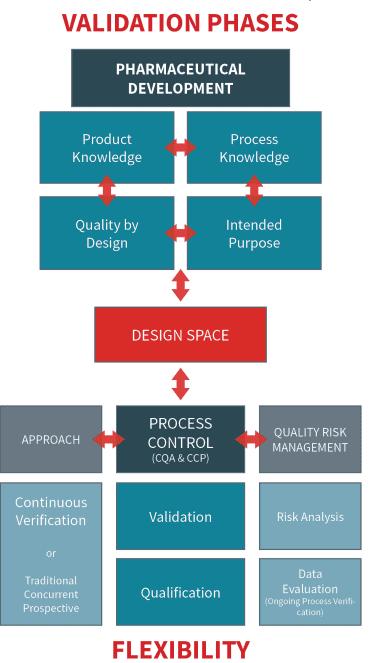
In today’s pharmaceutical environment, validation and qualification form the backbone of a scientifically sound and Quality by Design (QbD) approach. They provide the data and confidence needed to confirm that every aspect of your process—from equipment and personnel to utilities—consistently delivers product quality. This understanding defines your design space, the safe operational limits that ensure product integrity.
Stay within the design space, and you operate confidently. Move outside it, and you risk regulatory interventions and potential delays. That’s why QRM (Quality risk management) is crucial—it integrates scientific rationale with risk assessment to support robust, reproducible processes.
By qualifying systems and validating processes, you build a foundation for continuous monitoring, regulatory compliance, and long-term product integrity. This approach also aligns with the flexible, science- and risk-based regulatory frameworks encouraged in the updated Annex 1.
💡 Learn more about the latest Annex 1 updates and how to strengthen your contamination control strategy.




